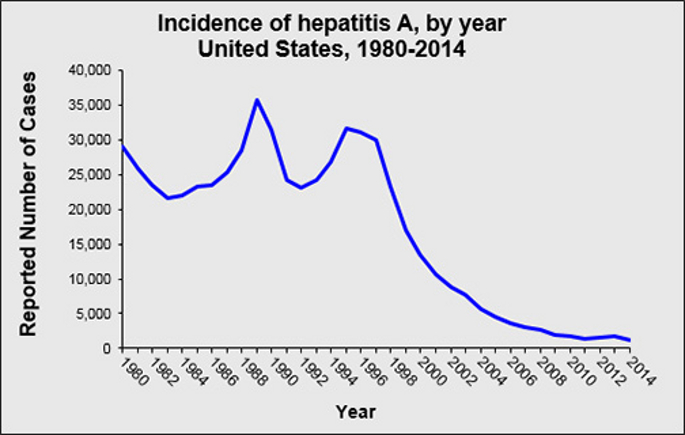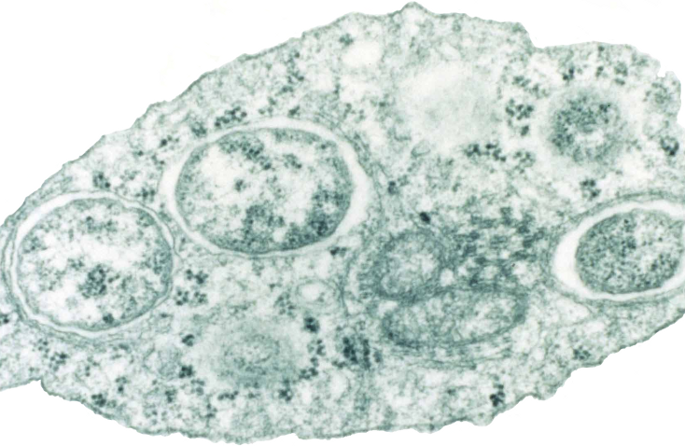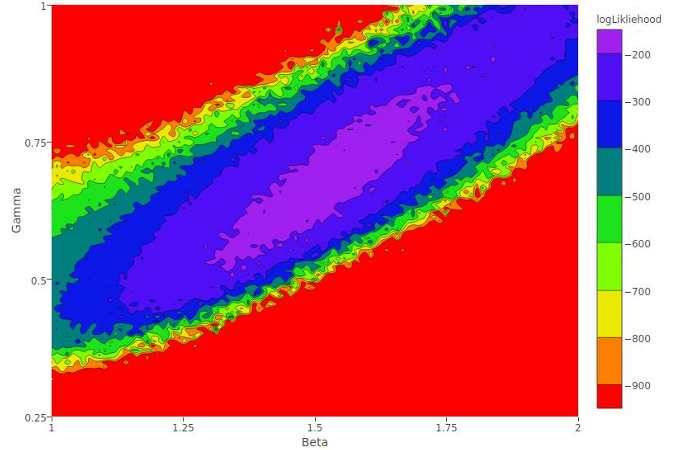
Natural history and genetic diversity of Dracunculus spp.
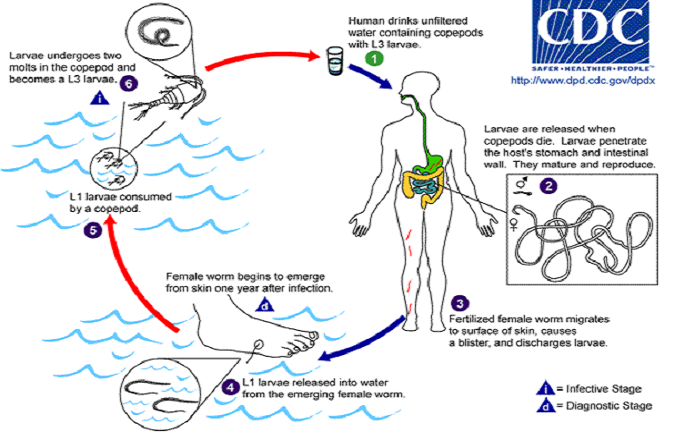
Alec Thompson, a Microbiology major from the University of Oklahoma, worked in the lab of Dr. Michael Yablsey to increase understanding of the genetic diversity of a parasite. Abstract: Dracunculus spp. are spiruroid nematode parasites that live in the subcutaneous tissues or abdominal cavity of mammals. The most important species is D. medinensis, the human