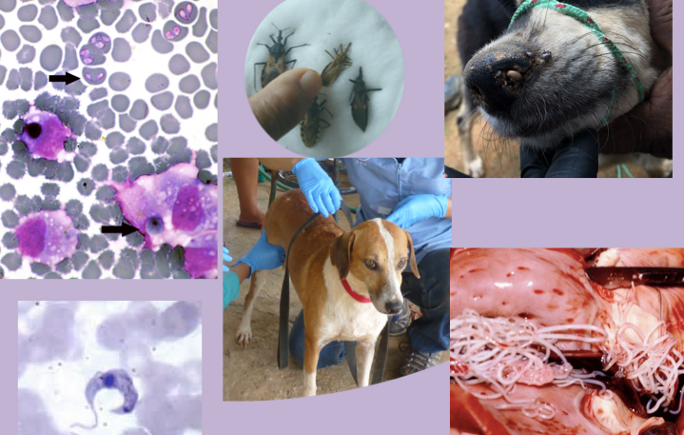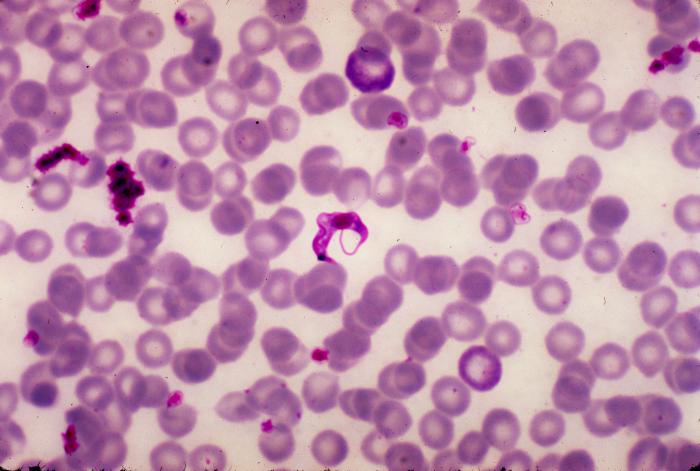
Associations between biotic and abiotic factors and Chagas disease vector abundance in palm trees across different habitat types
Jason Soriano, a freshman from University of California Berkeley, worked with Dr. Nicole Gottdenker and Christina Varian to investigate the factors influencing the abundance of an important disease vector. Abstract: In the Americas, an estimated 8 million people are infected with Chagas disease, a tropical, vector-borne infectious disease that can be life threatening if not